

治験への参加を考えたとき、あるいは家族が参加することになったとき、こんなふうに思ったことはないでしょうか。
「これって、本当に安全なの?」 「誰かがちゃんとチェックしてくれているの?」 「何かあったとき、自分は守ってもらえるの?」
当然の疑問です。治験は、まだ世の中に出ていない薬や治療法を人に試す行為である以上、不安を感じるのは当たり前のことです。
でも、ひとつ知っておいてほしいことがあります。治験の安全性を守るための仕組みは、制度として・法律として・そして「人」として、重ねて設計されています。
今回は、その中核を担う治験審査委員会(IRB:Institutional Review Board)の役割について解説します。IRBとは一言でいえば、「この治験、本当に人に行っていいの?」を第三者がきちんと確認する機関です。
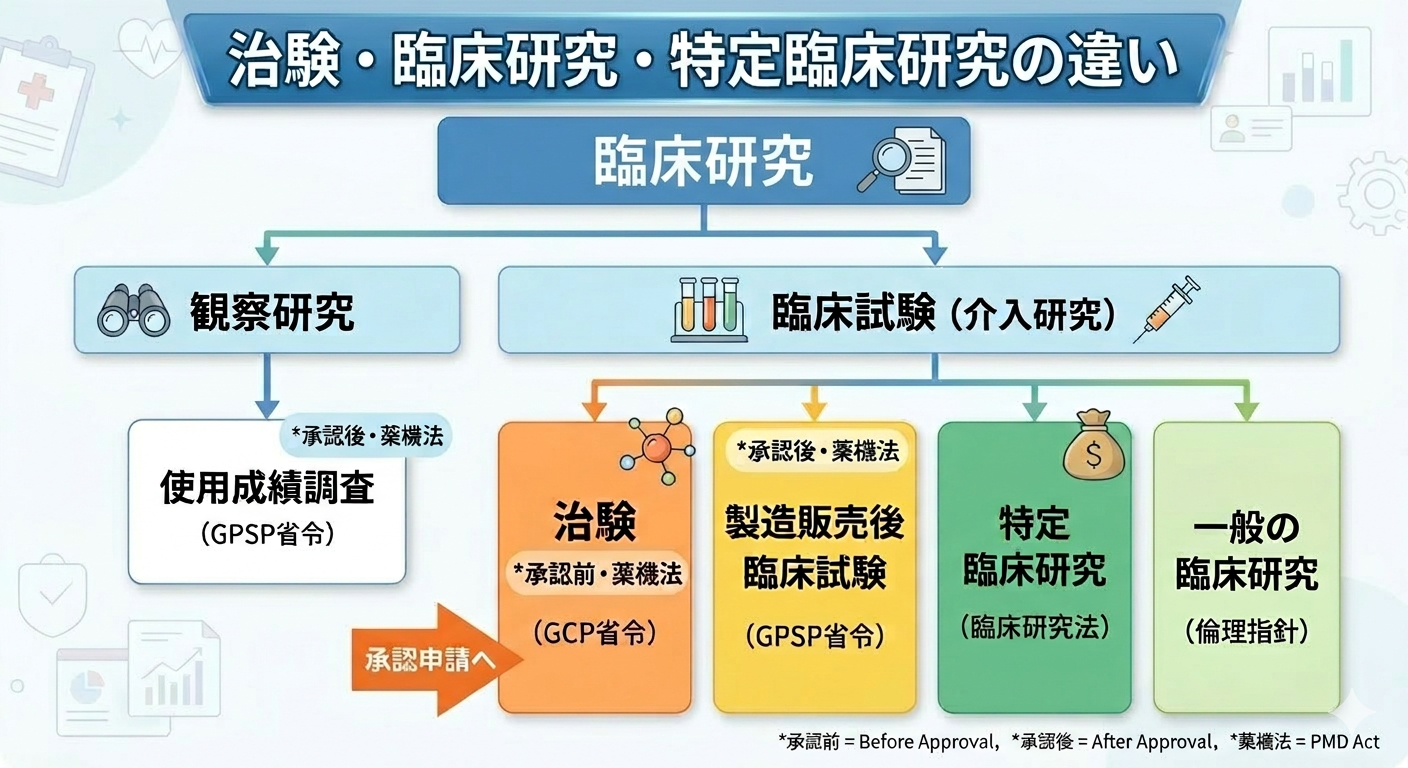
治験が始まる前に何が起きているのか
治験を実施したい製薬会社や研究者は、「やりたい」と思ってもすぐに患者さんへ声をかけることはできません。
まず治験実施計画書(プロトコル)を作成します。対象とする疾患、薬の種類と用量、参加者に生じうるリスク、緊急時の対応方法――これらを細かく文書化したものです。
そして、その計画書をIRBに提出し、審査を受けます。IRBが「倫理的に問題ない」「参加者へのリスクと得られる利益のバランスが妥当だ」と判断して初めて、治験は開始できます。IRBの承認なしに治験を一歩も進めることはできない、これが大前提です。
IRBはどんな人たちで構成されているのか
「治験審査委員会」と聞くと、医師ばかりが集まる専門家集団を想像するかもしれません。しかし実際は違います。
IRBのメンバー構成は、GCP省令(医薬品の臨床試験の実施の基準に関する省令)によって細かく定められています。医師・薬剤師・看護師などの医療専門家に加え、一般市民(非専門家)およびその医療機関と利害関係のない外部の人間を必ず含めなければなりません。
これは意図的な設計です。医療専門家だけが集まると「医学的に合理的かどうか」という視点が強くなりすぎる場合があります。そこに一般の視点・外部の客観的な視点を組み込むことで、「患者さんにとってどうか」「社会的に許容できるか」という問いが常に議論の場に持ち込まれます。
IRBは、治験の科学的妥当性だけでなく、参加者にとって倫理的に許容できるかどうかを判断する場所です。
IRBは何を審査しているのか
IRBは、GCP省令第三十二条に基づき、治験実施機関の長から審査を求められたとき、提出された資料をもとに文書で意見を述べる義務があります。その審査の核心は2点です。
「この治験は、倫理的・科学的に妥当か」 「この治験は、この医療機関で実施するのに適切か」
この2つの問いに答えることがIRBの本質的な役割です。審査の対象は多岐にわたりますが、参加を検討している方にとって特に身近な観点をいくつか紹介します。
リスクと利益のバランス
参加者に生じうるリスクの内容と程度を検討し、それが「新しい治療へのアクセス」「医学の進歩への貢献」といった利益と比較して受け入れられる範囲かどうかを審査します。
説明と同意(インフォームド・コンセント)のプロセス
参加者への説明文書(ICF:インフォームドコンセントフォーム)が、リスクも含めて正確に・理解できる言葉で記載されているかを確認します。特に「断ってもいい」「いつでも理由なくやめられる」という辞退の権利が、きちんと伝わる内容になっているかが重視されます。
治験関連業務に携わる立場から正直に言うと、ICFは細かい表現の一つひとつまで修正を求められることが珍しくありません。「この言い回しでは患者さんに誤解を与えないか」「専門用語が難しすぎないか」――そうした視点で何度も見直されます。
参加者の選び方の公正さ
特定の属性の人たちに負担が集中していないか、重篤な患者さん・子ども・判断能力が低下している方など、脆弱な立場の人が不当に参加させられていないかも審査されます。
IRBは承認するだけでなく、「この部分は修正が必要」と差し戻すこともあり、計画そのものを却下することもあります。
治験が始まった後も、IRBは機能し続ける
IRBの役割は、治験開始で終わりではありません。
実施中も定期的な進捗報告を受けて、「このまま続けていいか」を継続的に判断します。参加者に予期しない重篤な副作用が発生した場合は報告を受け、必要であれば治験の中断・中止を勧告する権限を持っています。
治験は始まったら最後まで止められない、ではないのです。開始後も、常に誰かが見守り続けています。
治験の安全性を支える多層的な仕組み
IRBは重要な仕組みのひとつですが、治験の安全性はそれだけで守られているわけではありません。
-
GCP(Good Clinical Practice)への準拠
日本で実施する治験はすべて、厚生労働省が定めるGCP省令に従って実施されなければなりません。GCPは、治験に関わる医療機関・製薬会社・すべての関係者が守るべきルールを体系的にまとめた基準です。 -
モニタリングによる継続的な品質管理
治験の実施中は、製薬会社の担当者(臨床開発モニター/CRA)による「モニタリング」が定期的に行われます。治験が計画書通りに正しく実施されているかを現場で確認するプロセスです。 -
副作用発生時の報告義務
参加者に重篤な副作用が発生した場合、規定の期間内に国への報告が義務づけられています。
IRBが「始める前の倫理的な審査」を担うとすれば、GCPとモニタリングは「進行中の品質と安全の管理」を担います。複数の仕組みが重なり合うことで、治験の安全性は守られています。
参加を検討しているなら、まずこの一言を
治験への参加を考えているなら、担当医師やCRCにこう尋ねてみてください。
「この治験は、IRBの承認を受けていますか?」
答えは必ずYESのはずです。承認を受けていない治験は実施できません。ただ、自分の言葉で確認することには意味があります。自分が参加しようとしているものが、正規の手続きを経ていると自分自身が知る――その一歩が、安心の土台になります。
また、参加の是非を含め、不安なことは遠慮なく担当医に相談してください。治験への参加は任意であり、参加しないことで通常の診療に影響が出ることはありません。
「守られる仕組みを知ること」は、参加を決める力にも、「今は参加しない」という判断を支える力にもなります。大切なのは、知らないまま決めないことだと思っています。
まとめ
-
IRB(治験審査委員会)は、「倫理的・科学的に妥当か」「この医療機関で実施するのに適切か」という2つの軸で治験を審査する第三者機関。リスクと利益のバランス、説明・同意のプロセス、参加者の選び方などはその代表的な確認事項です
-
IRBの承認なしに治験は実施できない
-
IRBの役割は治験開始後も継続し、中断・中止を勧告する権限を持つ
-
GCP省令・モニタリング・副作用報告義務など、治験の安全性は多層的な仕組みで守られている
治験は未知を扱う医療です。でも、無防備ではありません。あなたの見えないところで、たくさんの人と仕組みが重なって、参加者を守ろうとしています。そのことを知ったうえで、自分のペースで判断してほしいと思っています。

コメント